Jeg har uttrykt min beundring for de gamle fysikerne tidligere. En av mine favoritt-sitater er fra Richard Feynman: ”What I cannot create, I do not understand”. Det får meg til å tenke på at vi som jobber med å forstå biologiske systemer har en lang vei å gå. Og siden jeg er relativt ung så fyller den tanken meg med håp om fremtidige utfordringer og suksesser (Ja, jeg fyller 40 år i år; Og ja, jeg har litt krise; Nei, det er ikke irrasjonalt. Snarere tvert imot: jeg har brukt opp halve livet og om det ikke gir deg litt angst så mangler du enten ambisjoner eller så er du Superman; Nei, jeg er ikke Superman).
Feynmans sitat var tittelen på en Science kommentar like før jul; temaet var et nylig gjennombrudd innen proteindesign. Proteindesign? Et protein er en kjede av aminosyrer. Disse aminosyrenes rekkefølge og (fysiokjemiske) egenskaper bestemmer proteinets form (også kalt struktur eller fold). Og et proteins form er helt avgjørende for dens funksjon; akkurat som en hammer er formet slik at den egner seg til å slå inn spiker så har hemoglobin evolvert en form som egner seg for å transportere oksygen. Helt siden Anfinsen formulerte den termodynamiske hypotesen, har protein-folding-problemet vært en magnet på alle oss som liker å studere biologi ved hjelp av datamaskiner (bioinformatikk). Hypotesen sier at et proteins form utelukkende er bestemt av dens aminosyresekvens og problemet er å lage en datamaskin som tar alle genene vi har sekvensert og spytter ut perfekte modeller av hvordan de korresponderende proteinene ser ut. Hadde vi hatt en slik datamaskin så ville det ha vært til stor hjelp i arbeidet med å, for eksempel, designe nye enzymer som effektivt konverterer biomasse til biodiesel. Så hva stopper oss?
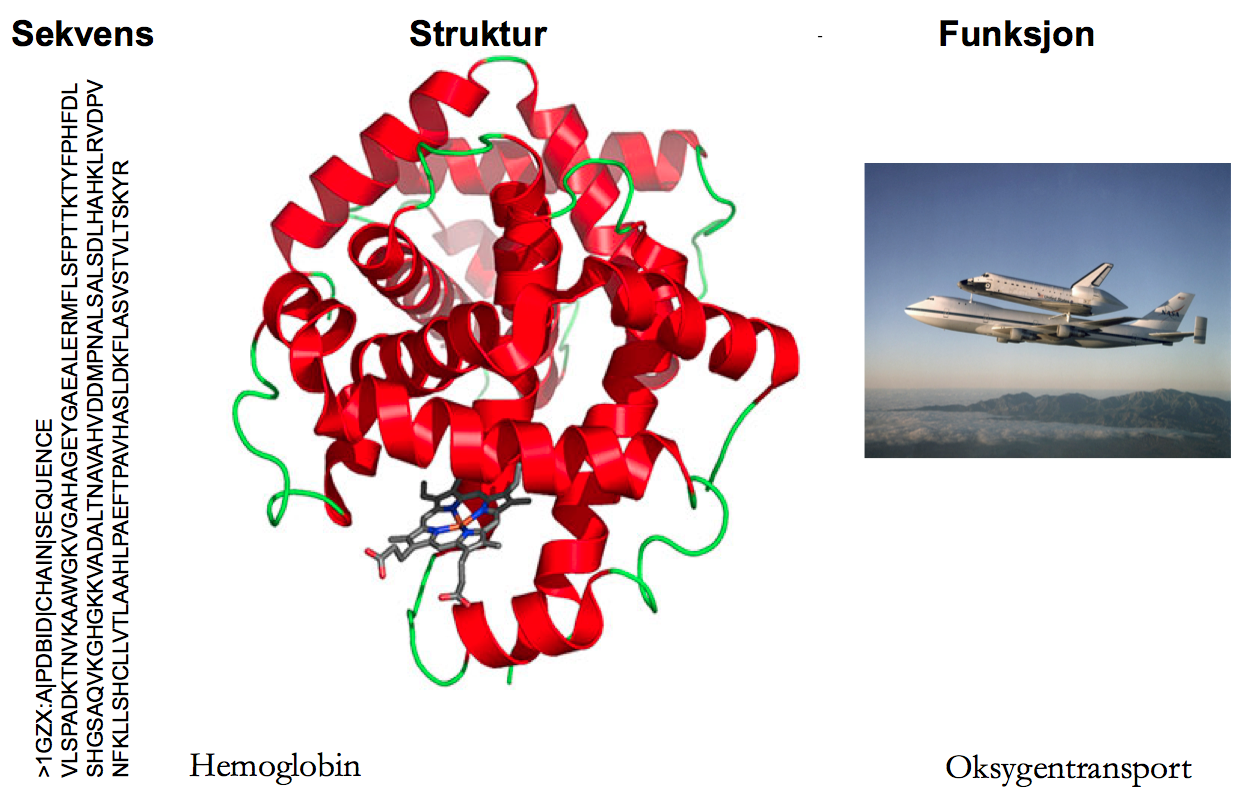
Datamaskiner løser problemer ved hjelp av søk: algoritmen søker etter den beste løsningen på et problem i rommet av alle mulige løsninger (søkerommet). Løsningene i vårt tilfelle er alle mulige måter et protein kan foldes på og den beste løsningen er den folden med lavest fri energi. Fri energi kan regnes ut ved hjelp av kvantemekanikk; det er ikke problemet her. Problemet er at et protein kan foldes på så uhorvelig mange ulike måter (Levinthal paradox): et relativt lite protein på 100 aminosyrer kan foldes på 10^143 ulike måter (til sammenligning er det bare 10^80 partikler i universet). Bare en av disse foldene er korrekt; de andre ville kunne lede til et protein som ikke gjør det det skal. Akkumulering av slike feilfoldede proteiner (forårsaket av mutasjoner) er årsaken til flere sykdommer som for eksempel Alzheimer.
Annethvert år holdes det en konkurranse der forskere kan sende inn forslag på strukturen til proteiner der denne ennå ikke er kjent eksperimentelt. Kort fortalte er ingen i nærheten av å ha løst protein-folding-problemet. Så hvorfor finner vi ikke formen til alle proteiner ved hjelp av eksperimenter? Fordi dette er en dyr og tidkrevende prosess (det finnes opp mot 20 millioner proteinsekvenser i forskerverdenens databaser, men bare rundt 100 000 proteinstrukturer). Og fordi … det jeg ikke kan bygge selv, har jeg heller ikke forstått. Vi vil designe nye proteiner og da må vi forstå protein folding.
NB: Stemmer Anfinsens termodynamiske hypotesen? Tja, det diskuteres fortsatt, men det finnes definitivt unntak.

